Learning the structural diversity in random protein sequence space
Filip Buchel1,2†, Tereza Neuwirthova1†, Theodora Tureckiova1, Gustavo Fuertes3, Ales Benda6, Dalibor Panek6, Matus Fricek1, Mohammed AlQuraishi4*, Klara Hlouchova1,5*
1Department of Cell Biology, Faculty of Science, Charles University, Prague, Czech Republic 2Department of Biochemistry, Faculty of Science, Charles University, Prague, Czech Republic 3Institute of Biotechnology of the Czech Academy of Sciences, Vestec, Czech Republic 4Department of Systems Biology, Columbia University, New York, NY, USA 5Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague, Czech Republic 6Imaging Methods Core Facility at BIOCEV, Vestec, Czech Republic
†These authors contributed equally to this work *Correspondence to: m.alquraishi@columbia.edu , klara.hlouchova@natur.cuni.cz
Abstract
The universe of possible protein sequences is astronomically large, yet our understanding of the sequence-structure relationship is confined to the infinitesimal fraction used currently by life. Determining whether “foldable” architectures are rare singularities or accessible solutions is critical for understanding protein evolution and designing novel proteins. Here, we map the structural landscape of random sequence space by screening one million synthetic proteins using a high-throughput in vivo FRET biosensor. We reveal that this space is structurally heterogeneous, populated not only by disordered chains and stress-inducing aggregates but also by “benign” compact structures that resemble globular proteins and evade cellular chaperone responses. By training machine learning models on these phenotypes, we show that structural potential is learnable and generalizes to natural proteomes. These findings demonstrate that biology-like folds are accessible from random sequences with surprising frequency, providing data required to expand generative protein design beyond evolutionary priors.
Introduction
Protein sequences occupy an astronomically large combinatorial space, and learning to navigate this space is a central challenge in protein design. Most contemporary advances in structure prediction and de novo protein design rely on naturally occurring sequences produced by evolutionary exploration of sequence space [1–4]. However, available sequence annotations and experimentally solved structures are unevenly distributed, with strong biases toward a limited set of organisms and protein families [5]. As a result, large regions of sequence space - used by biology, evolutionary dormant or entirely unexplored - remain sparsely sampled, potentially limiting the generality of current design principles.
Random sampling of protein sequence space provides a complementary, orthogonal strategy for exploring structure-function relationships. Selections from random sequence libraries have yielded proteins with diverse functions, including ligand binding and enzymatic activity [6–8]. Random polypeptide libraries have been used to train predictors of aggregation propensity, underscoring the potential of probing global sequence-structure relationships beyond evolutionary constraints [9]. Finally, Tretyachenko and colleagues employed random libraries with defined amino acid compositions to separate populations with distinct solubility and structural content using proteolytic resistance assays [10]. While these experiments revealed coarse-grained differences across sequence space, they lacked the resolution to systematically map the diversity of structural properties at scale.
Here, we systematically characterize the structural properties of a large random protein sequence library using an in vivo FRET-based proximity biosensor [11][12]. After validating the ability of the assay to resolve folding and oligomeric states using control proteins, we apply it to high-throughput screening of over one million random sequences, revealing discrete populations with distinct structural and aggregation phenotypes. We further show that sequence-derived features enable training of a neural network classifier that assigns individual sequences to FRET-defined categories. Together, these results demonstrate how unbiased sampling of random sequence space can complement evolutionary datasets and help mitigate sparse-sampling biases in protein design.
Results
High-throughput FRET screening resolves structural diversity within a random sequence library
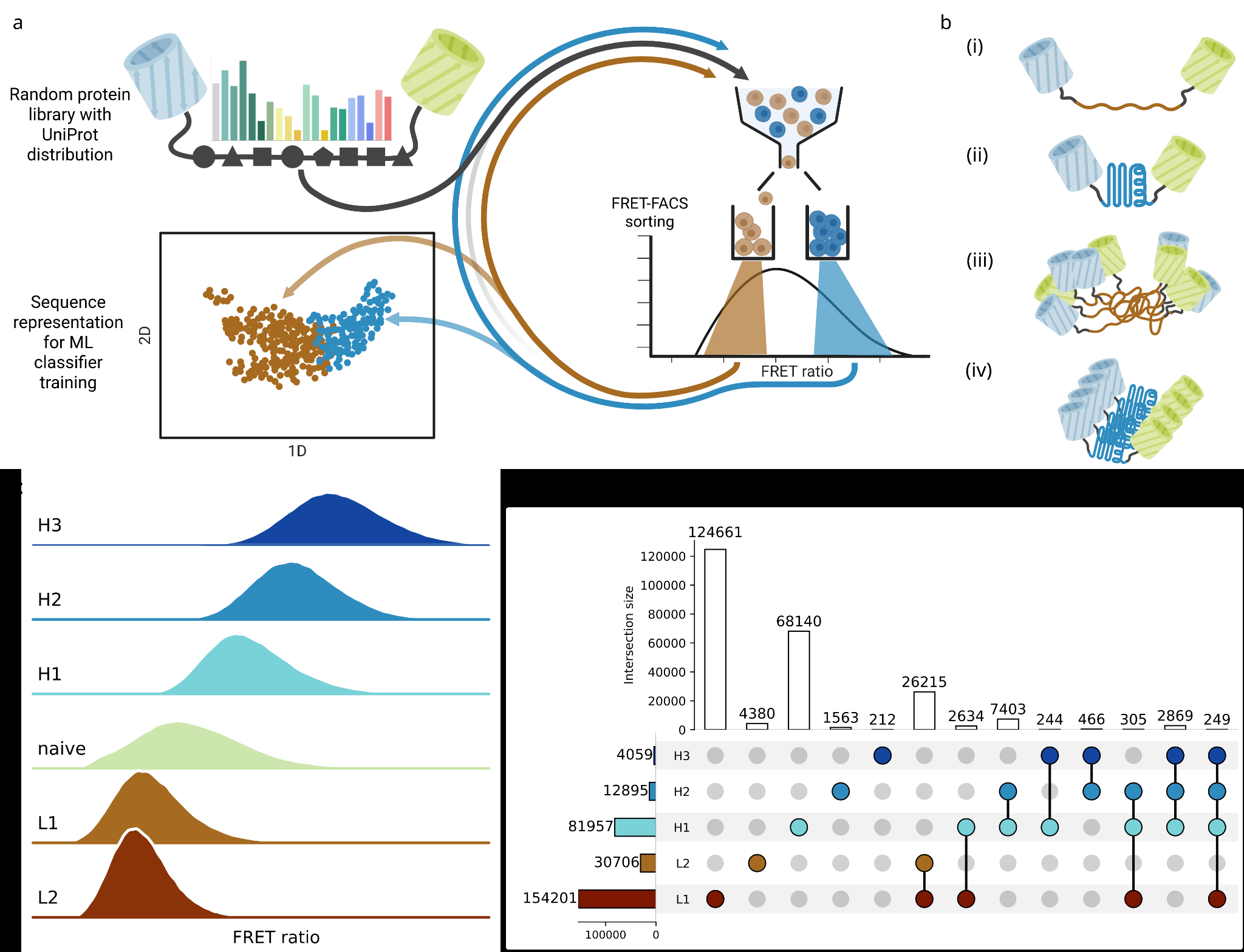
To sample the sequence space of small proteins, we generated a library with 10^9 randomized positions with a defined amino acid composition. The library sequences were designed using the CoLiDe algorithm, previously applied to bulk studies of large random libraries with defined compositional constraints [10][13]. To enable efficient library construction at this scale, we developed a Golden Gate Assembly-based strategy that builds the library from two oligonucleotides while preserving full diversity at the ligation junction. This approach allows cost-effective, PCR-free library generation (Methods). Using an amino acid distribution reflecting average UniProt frequencies [10], we constructed a library of approximately four million unique variants, hereafter referred to as 20F109. To test if the sequences encoded in this library are distinct from those used in nature, we compared the 20F109 subpopulations to length-matched proteins from four natural kingdoms using embedding projections. Despite adhering to natural amino acid frequencies, the library sequences clustered on the opposite side of the embedding space from natural proteins (Supplementary Figure 5d), confirming that our library represents a unique part of sequence space distinct from that currently explored by biology.
Next, we established a biosensor capable of reporting on both the internal structure and intermolecular association in a high-throughput manner. The proteins were expressed in E. coli as fusions to a genetically encoded FRET pair, such that FRET efficiency reports on the proximity of the fluorophores, defined by structural and oligomeric properties of the insert (Figure 1b). We tested the system using a panel of controls, including glycine-serine repeat (GS) linkers of varying length, length-matched globular E. coli proteins, and previously characterized random proteins [14]. Characterization of the system was performed on multiple levels: in vitro measurements of cell lysates using time-resolved fluorescence spectroscopy; in vivo measurements by fluorescence lifetime imaging microscopy (FLIM) to assess localization (quantified by mean photon arrival time) and flow cytometry with the FRET ratio read-out (ratio of FRET channel to donor channel intensity; for details see Methods) to simulate the high-throughput regime. As expected, FRET efficiency decreased with increasing GS linker length and displayed uniform intracellular distributions [15]. In contrast, for globular E. coli proteins, FRET efficiency did not correlate with protein length; instead, distinct efficiencies were observed for proteins of similar size (for example, IscA, BolA, and CspA; Supplementary Figure 1b), consistent with differences in their conformational properties.
We next examined a subset of control proteins from Tretyachenko et al. [14], as those random sequences closely resemble variants in the 20F109 library. To illustrate the range of the FRET signal, we selected three representative controls: the disordered protein 665 and the structurally enriched proteins 1259 and 1856. Protein 665 expressed in a fully soluble form, supported by dispersed FLIM signal and FRET efficiency consistent with an unstructured polypeptide (Supplementary Figures 1 and 2). In contrast, proteins 1259 and 1856 displayed limited solubility (Supplementary Figure 1), in accordance with previous observations of increased aggregation propensity in random protein sequences with high structural content [14][16]. Despite similar localization to inclusion bodies observed by FLIM, proteins 1259 and 1856 exhibited markedly different FRET efficiencies in both in vivo and in vitro measurements (Supplementary Figures 1 and 2), possibly suggesting distinct aggregation states. Indeed, FRET-based biosensors have previously been used to resolve structural heterogeneity and packing density within inclusion bodies [17][18]. Under this framework, the low FRET signal of protein 1259 suggests a low-density aggregate, whereas the high FRET efficiency of protein 1856 indicates a more compact tightly packed assembly, following the model proposed in Figure 1b (panels iii and iv). Together, these results demonstrate that the FRET-based assay can resolve distinct structural and oligomeric states of random proteins.
Encouraged by the performance of the system, we proceeded with applying the assay in a high-throughput regime to screen the 20F109 library. The subcloned library with an initially estimated diversity of 3-5 million members was FACS-sorted for FRET ratio signal in multiple rounds. To enrich sequences with the most extreme signal, we gated shoulders of the FRET ratio distribution (roughly 10 %) in 2 rounds for low FRET (L1 and L2) and 3 rounds for high FRET path (H1, H2 and H3). Progressive shifts in FRET ratios were observed in the recovered subpopulations, reflecting successful enrichment of structurally distinct species (Figure 1b). Following the sorting, all subpopulations were submitted to amplicon NGS to identify sequence features underlying the observed FRET signal variations.
Hydrophobicity and compactness drive FRET-defined protein states
We first generated a reference database by clustering the naive library pool at 99% sequence identity, yielding approximately 4 million variants. Sequencing reads from the sorted subpopulations were mapped to the reference database to quantify the abundance changes across successive sorting rounds. Approximately one quarter of the naive library members were recovered in at least one selection round, with diversity progressively decreasing in later rounds, as expected (Figure 1c).
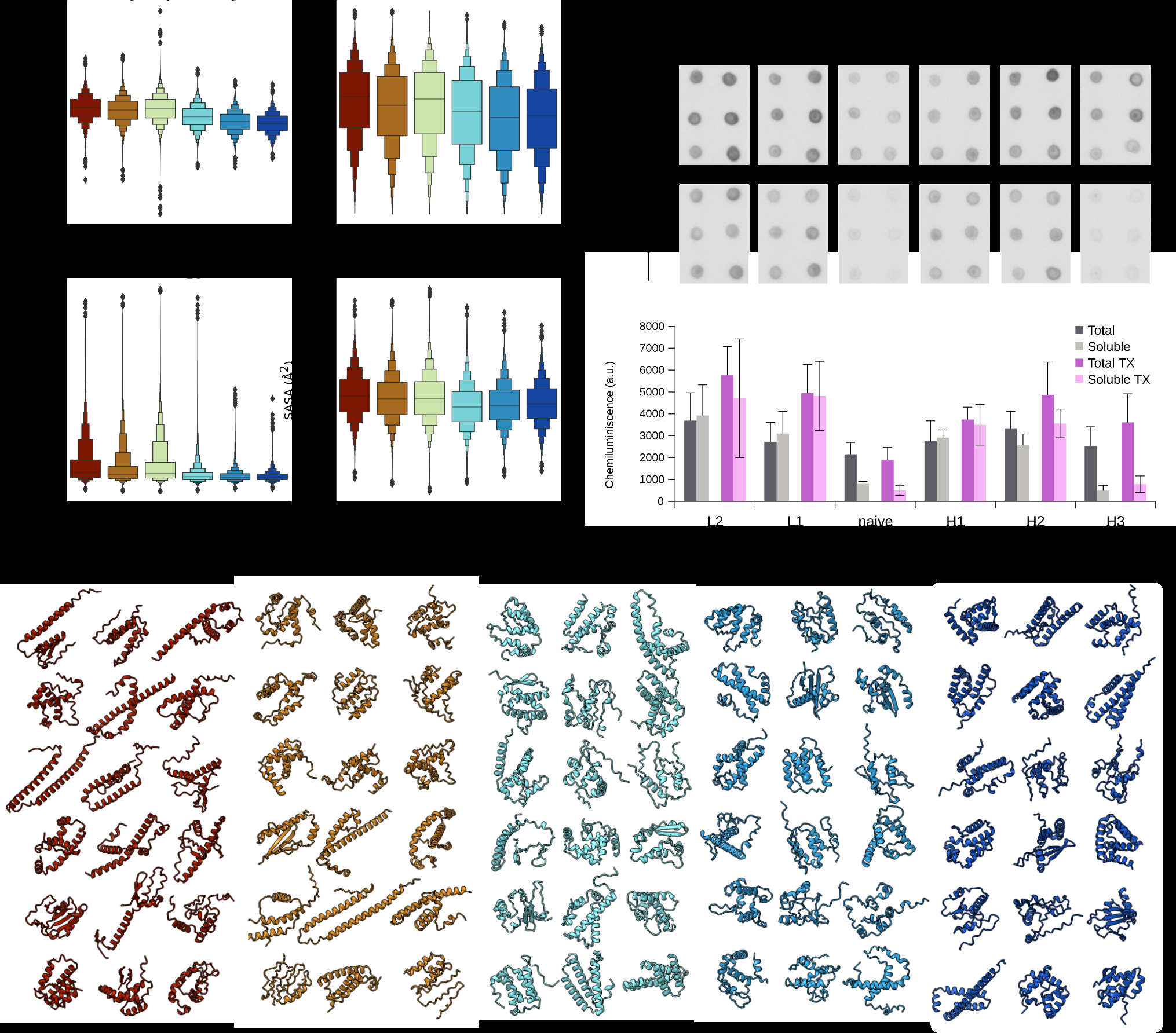
To identify sequence and structural features associated with the observed FRET phenotypes, we extracted a set of primary sequence descriptors (composition, hydrophobicity, isoelectric point, and sequence complexity), together with features derived from computationally predicted structures by ESMFold [2] (secondary structure content, radius of gyration, and solvent accessible surface area). (Figure 2a). A prominent trend was observed in hydrophobicity, gradually decreasing towards the high-FRET population (H3), accompanied by enrichment of charged residues (arginine, glutamate, and aspartate) (Figure 2a, Supplementary Figure 3a). Structural predictions further indicated that low-FRET subpopulations (L1 and L2) were enriched in extended, predominantly helical conformations with larger radii of gyration and greater solvent exposure, whereas high-FRET populations (H1-H3) were characterized by more compact structures with increased beta-sheet content (Figure 2a and Supplementary Figure 3b). Collectively, the extracted features show a smooth transition from extended, helical structures in low-FRET variants to more hydrophilic sequences adopting compact, globular folds with increased secondary structure and topological diversity in high-FRET variants.
To validate the screening results, we selected high-abundance candidates from each sorting round for individual characterisation. Flow cytometric analysis showed that the selected variants spanned nearly the full range of FRET ratios observed in the naive library, whereas FLIM data revealed substantial heterogeneity within the low-FRET populations. All high-FRET candidates exhibited localized intracellular signals with short donor lifetimes, consistent with the behaviour of the control protein 1856. In contrast, low-FRET candidates comprised two distinct phenotypic classes: variants displaying diffuse intracellular fluorescence (comparable to largely unstructured proteins), and variants with localized low FRET signals, indicative of loosely packed aggregates (Supplementary figure 2a).
To further distinguish between these aggregation modes, we assessed activation of the ibpA promoter in response to expression of selected candidates [19]. Both the aggregation-prone control protein 1259 and the tested low-FRET candidates with localized expression induced a chaperone response via ibpA promoter activation, whereas no detectable response was observed for unstructured variants, well-folded proteins, or the tested high-FRET candidates (Supplementary figure 2c).
Together, these analyses resolve multiple, mechanistically distinct structural and aggregation states within the random sequence library, establishing a framework in which compact, folded protein-like architectures can be distinguished from unstructured and aggregation-prone species and systematically followed across sequence space.
Biology-like protein folds emerge and can be traced across sequence space
To assess the solubility of sequences enriched by FRET-based sorting, we expressed selected variants without the FRET reporters in an E. coli-based reconstituted cell-free system. Coding sequences were excised from the pETMF vector and expressed with a C-terminal FLAG tag, and expression was evaluated with a dot-blot assay (Methods). Relative to the naive library, all FRET-sorted subpopulations exhibited increased expression yields, although the size difference between the naive and sorted libraries might be a contributing factor. Triton X-100 generally improves expression yields of all sorted subpopulations, with the most pronounced effect in L1 and L2, potentially stemming from increased hydrophobicity of these subpopulations. Low-FRET subpopulations were enriched in soluble variants, whereas the fraction of soluble proteins decreased progressively across successive high-FRET selection rounds (Figure 2b).
To visualize the structural variability captured by FRET-based sorting, we selected 18 representative sequences from each sorted subpopulation and examined their previously generated ESMFold structural models, prioritizing highly abundant variants (Figure 2c). Consistent with trends inferred from sequence- and structure-derived metrics (Figure 2a), low-FRET variants predominantly adopt extended conformations enriched in alpha-helical elements. In contrast, high-FRET variants exhibit increased turn content and overall structural compaction, yielding architectures reminiscent of globular proteins (Figure 2a, 2c, and Supplementary Figure 3c). Within these high-FRET subpopulations, we observed a range of biology-like folds, including helical bundles and simple α/β structures spanning multiple topologies (Figure 2c).
Across successive rounds of high-FRET selection, the radius of gyration and solvent-accessible surface area of the predicted structures remained largely unchanged, indicating sustained structural compactness. However, hydrophobicity and helical content decreased progressively from the first to the third high-FRET round (H1 to H3; Figure 2a), a trend that coincided with reduced solubility, as observed in cell-free expression assays (Figure 2b). Together, these results suggest that increasing selection stringency favors compactness per se, but progressively enriches aggregation-prone variants, whereas compact, biology-like folds of diverse topologies are preferentially recovered in the earlier high-FRET selection rounds.
Learning the structural states captured by the latent sequence representation
A central goal of protein design is not only to characterize individual regions of sequence space, but to learn representations that enable navigation across this space independently of specific experimental assays or libraries. If structural and aggregation states correspond to intrinsic, sequence-encoded features, then they should be detectable directly from sequence, without explicit knowledge of experimental labels or structural annotations. We therefore asked whether the structural states resolved by FRET-based selection leave identifiable signatures in the primary sequence that can be captured by modern protein language models.
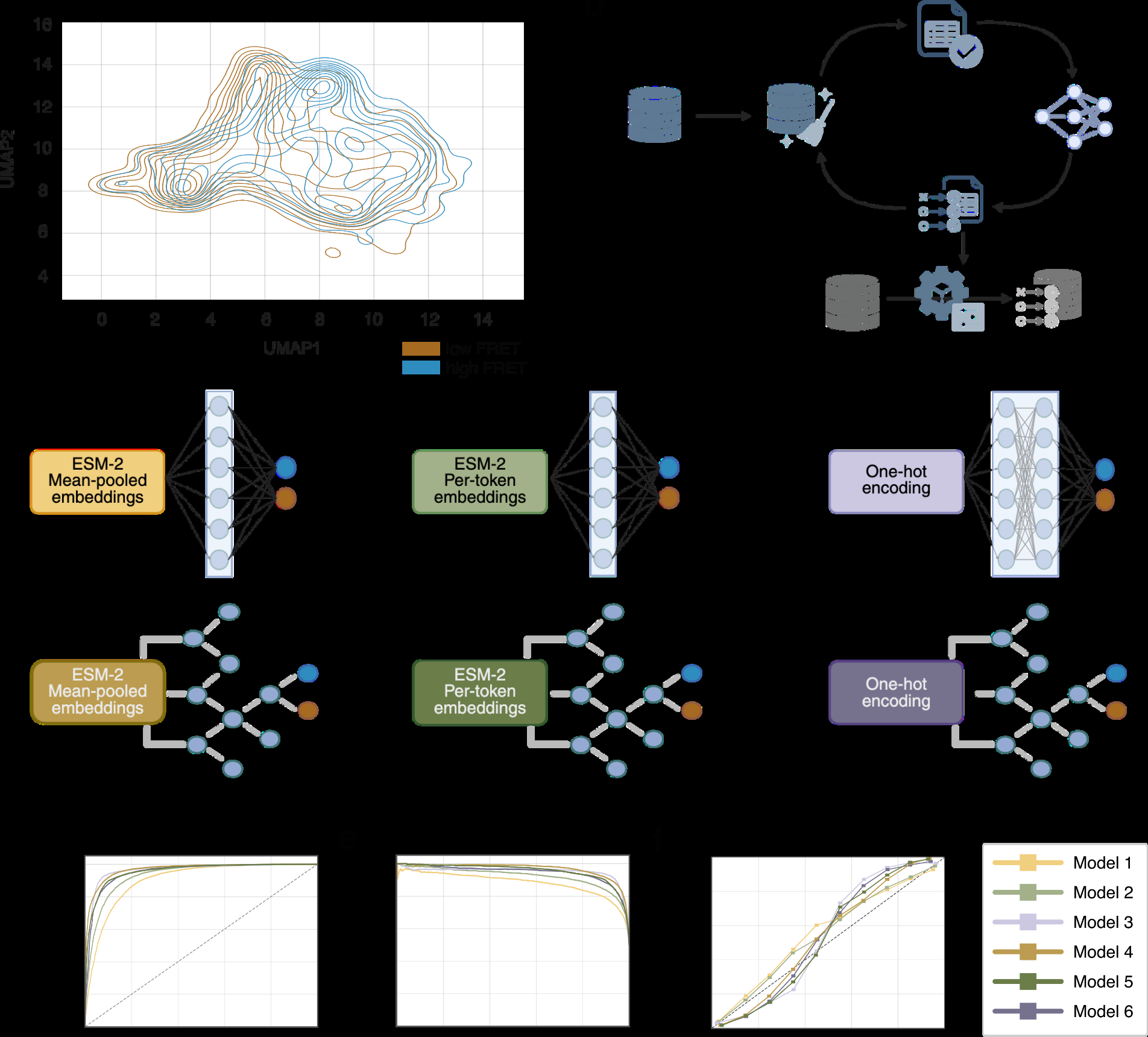
To address this, we projected sequences from low- and high-FRET subpopulations into a 2D embedding space derived from the pretrained ESM-2 model [2]. Despite the absence of supervision, sequences from the two subpopulations occupied partially distinct regions of the embedding space (Figure 3a), indicating that language model representations capture the sequence features associated with the experimentally defined structural phenotypes. Clearest separation between the two subpopulations in 2D space was observed with mean embeddings projected with UMAP.
Encouraged by these results, we trained supervised binary classifiers to predict FRET-defined structural classes directly from sequence. Before training, sequences were filtered to remove duplicates between subpopulations and encoded using a sequence-representation strategy (Methods). We evaluated six distinct classifier architectures, ranging from random forests to deep neuronal networks, using different encoding strategies (Figure 3b and 3c), Given the overlap observed between different sorting rounds in the 2D embedding space (Supplementary figure 4f and 4g), sequences from all rounds were pooled for training (Methods). To mitigate overfitting and prevent data leakage, we employed an MMseqs2-guided diversity strategy for our train/test split, as proposed by Bushuiev & Bushuiev [20][21]. Specifically, we performed an all-vs-all sequence comparison (Methods) and withheld the most distant 20% of sequence variants from each class to serve as an independent test set.
All evaluated models showed strong classification performance, with high ROC-AUC and average precision values overall, confirming robust discrimination between classes (Figure 3d & 3e), Supplementary Table 1), even though not all used sequence encoding strategies were able to distinguish between the two subpopulations with the same level of sensitivity when visualized in 2D with PCA or UMAP (Supplementary Figure 4a-e). Among them, the Model 4 (mean ESM-2 embeddings + random forest) performed best globally (AUC = 0.97, AP = 0.97). Threshold-dependent analyses (Methods) further indicated that the best operating points clustered around 0.4-0.5, highlighting that performance differences between models are most relevant when selecting deployment thresholds and calibration requirements rather than in raw accuracy alone.
Reliability-diagram analysis (Figure 3f) further indicated model-specific calibration differences: Models 1-3 were generally closer to the diagonal (better calibrated, with mild underconfidence), whereas Models 4-6 showed stronger calibration drift, being overconfident at low-to-intermediate predicted probabilities and underconfident at higher probabilities. For additional analyses, Model 1 and Model 2 were selected as representative models because of similar classification performance of the models and best looking reliability curves of these two.
To further validate the classifier, we withheld an independent set of sequences from training. Specifically, the 100 most abundant sequences from each selection round (together with the 100 most abundant sequences from the pre-sorted library) were excluded prior to model training, and duplicate sequences were removed, yielding a total of 504 previously unseen variants. When evaluated on this held-out set, predictions from the Model 2 exhibited a systematic dependence on selection round: mean predicted probabilities for the high-FRET class increased progressively across successive high-FRET rounds, whereas probabilities for the low-FRET class decreased correspondingly across low-FRET rounds (Supplementary table 2). These trends indicate that the classifier captures sequence features correlated with the degree of enrichment during FRET-based selection, rather than merely separating binary classes.
To examine whether the classification logic extends beyond random sequences, we applied the trained model to natural proteins with diverse structural topologies. Proteins of length comparable to the random library were selected from UniProt [22] representing globular, coiled-coil, and transmembrane proteins classes, and from DisProt [23] to represent intrinsically disordered proteins (IDPs) (Methods). Sequences were classified using Model 1 (Supplementary figure 5a,b). Globular proteins were distributed relatively evenly between high- and low-FRET classes, whereas coiled-coil proteins showed a modest enrichment toward the high-FRET class. In contrast, IDPs were preferentially classified as low-FRET, and transmembrane proteins were predominantly assigned to the low-FRET class. Based on the models from each topology class with highest and lowest probability we hypothesize that proteins that have less compact structure and/or large proportion of exposed hydrophobic residues on the surface are preferentially assigned lower probability (classified low-FRET). Overall, these results indicate that the classifier, trained exclusively on random sequences, captures sequence features that generalize to natural proteins and align with known structural and solubility properties.
Discussion
A central question in evolutionary biology and protein biophysics is to what extent the structural diversity observed in nature reflects the intrinsic physical properties of polypeptides or rather represents the product of evolutionary constraints [24]. While the combinatorial vastness of sequence space implies that “foldable” sequences represent sparse, isolated solutions to the geometric problem of polypeptide packing, theoretical and sparse experimental sampling suggests otherwise. The field of statistical physics implies that polymer behavior, including the main elements of globular proteins - helices, sheets and hydrophobic cores - arise as emergent properties of random heteropolymers [25–27]. This view has been supported by sparse and coarse-grained experimental sampling that detected stable and compact structures within random sequence space [10][14][28]. Although the ease with which structure emerges from sequence space likely differs among folds, these studies have supported the view that evolution “edits” the inherent structure forms (e.g. for specific functions and stability properties) rather than creating new folds de novo [25][29]. However, bridging the gap between these observations and systems level data has remained a challenge. In this study, we systematically mapped the structural properties of random sequence space, moving beyond binary classifications of solubility to better resolve a continuum of conformational states. Previous efforts to probe this space have relied largely on survival-based assays, such as resistance to proteolysis or aggregation reporters [9][10]. While informative, these methods often blur the distinctions between solubility and folding or folding and aggregation and cannot reliably separate individual conformational states. By employing a high-throughput in vivo FRET biosensor, we successfully decoupled these properties, identifying discrete populations ranging from extended random coils to collapsed globular structures and aggregated assemblies. Strikingly, we observed that while increasing hydrophobicity generally drives compaction, it appears to separate the population into two distinct phenotypes: amorphous aggregates triggering chaperone responses, and compact, “biology-like” architectures that evade cellular quality control.
Our results challenge the assumption that high structural content in random sequences is inevitably linked to “toxic” aggregation. High-FRET variants enriched in our screen displayed structural compaction and increased secondary structure content reminiscent of globular proteins, yet many did not trigger upregulation of the ibpA stress response. This behavior parallels the recently described phenomenon of protein “agglomerates” - assemblies that are mechanically stable and compact but lack the exposed hydrophobic patches that typically recruit chaperones [30]. This distinction is vital for protein design: it suggests that the primary challenge in navigating random space is not merely finding sequences that collapse, but distinguishing between “benign” compactness (potential proto-folds) and aggregation. Our screen provides an experimental filter to isolate the former.
Interestingly, our data highlights the current limitations of homology-dependent structure prediction in unexplored regions of sequence space. While AlphaFold2 [31] and ESMFold [2] have revolutionized structural biology, they rely on co-evolutionary signals and known structural priors. As demonstrated in the study comparing the prediction accuracy of AlphaFold and ESMFold for monomeric and dimeric proteins, nearly half of the proteins that were incorrectly predicted by ESMFold and had low pLDDT values also lacked homologs [32]. Consequently, ESMFold assigned low confidence scores (pLDDT < 50) to our random sequences. While the resulting models often appear structured and our FRET data confirm compactness, the low pLDDT values indicate that these predictions cannot be definitively validated. Our results demonstrate that despite these low confidence scores, random sequences seem to contain learnable structural features that correlate with actual experimental sorting done with the FRET assay. By training a classifier on FRET-defined phenotypes, we show that neural networks can learn to distinguish structural states directly from sequence, even in the absence of evolutionary history. The classifier’s ability to generalize to natural proteins - correctly associating transmembrane domains with low-FRET (extended/membrane-integrated) and coiled-coils with high-FRET phenotypes - validates that the principles governing random and biological protein folding are convergent.
We acknowledge three specific limitations in our current approach. First, our library design sampled sequences with average amino acid distributions resembling natural proteins. Thus, the vastness of purely unbiased sequence space remains largely unexplored. Within this sampled space, we prioritized breadth over depth: by maximizing the number of unique variants rather than sequencing replicates, we accepted a degree of statistical noise in exchange for coverage. Consequently, while our filtering was robust, false positives may persist, and we cannot currently distinguish sequences that were truly unexpressable from those merely missed by the gating window. Second, while FRET effectively reports on compaction, it is a low-resolution proxy. The current setup does not fully resolve heterogeneity within the low-FRET population - which likely contains a mix of extended soluble chains and amorphous aggregates - a distinction that future experiments utilizing imaging flow cytometry could resolve. Finally, the “black box” nature of our classifier highlights an unsurprising knowledge gap. While the neural network effectively sorts phenotypes, the specific hidden traits it recognizes remain to be deciphered. Moving forward, the “proto-folds” identified here must be experimentally validated by high-resolution structural biology.
This work establishes a new high-throughput approach for exploring structural properties of vast sequence space. By generating structural categories for non-evolutionary sequences, we sample the missing “negative space” data required to train the next generation of AI models. Such datasets could be instrumental in refining algorithms to recognize folding propensity based on first principles rather than homology, ultimately enabling the design of novel protein folds that lie beyond the scope of evolution.
References
- Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
- Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
- Dauparas, J. et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022).
- Ferruz, N., Schmidt, S. & Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 13, 4348 (2022).
- Ding, F. & Steinhardt, J. Protein language models are biased by unequal sequence sampling across the tree of life. 2024.03.07.584001 Preprint at https://doi.org/10.1101/2024.03.07.584001 (2024).
- Keefe, A. D. & Szostak, J. W. Functional proteins from a random-sequence library. Nature 410, 715–718 (2001).
- Frumkin, I. & Laub, M. T. Selection of a de novo gene that can promote survival of Escherichia coli by modulating protein homeostasis pathways. Nat. Ecol. Evol. 7, 2067–2079 (2023).
- Schnettler, J. D. et al. Selection of a promiscuous minimalist cAMP phosphodiesterase from a library of de novo designed proteins. Nat. Chem. 16, 1200–1208 (2024).
- Thompson, M. et al. Massive experimental quantification allows interpretable deep learning of protein aggregation. Sci. Adv. 11, eadt5111 (2025).
- Tretyachenko, V. et al. Modern and prebiotic amino acids support distinct structural profiles in proteins. Open Biol. 12, 220040 (2022).
- Philipps, B., Hennecke, J. & Glockshuber, R. FRET-based in Vivo Screening for Protein Folding and Increased Protein Stability. J. Mol. Biol. 327, 239–249 (2003).
- Aubel, M. et al. High-throughput Selection of Human de novo-emerged sORFs with High Folding Potential. Genome Biol. Evol. 16, evae069 (2024).
- Tretyachenko, V., Voráček, V., Souček, R., Fujishima, K. & Hlouchová, K. CoLiDe: Combinatorial Library Design tool for probing protein sequence space. Bioinformatics 37, 482–489 (2021).
- Tretyachenko, V. et al. Random protein sequences can form defined secondary structures and are well-tolerated in vivo. Sci. Rep. 7, 15449 (2017).
- Evers, T. H., van Dongen, E. M. W. M., Faesen, A. C., Meijer, E. W. & Merkx, M. Quantitative Understanding of the Energy Transfer between Fluorescent Proteins Connected via Flexible Peptide Linkers. Biochemistry 45, 13183–13192 (2006).
- Heames, B. et al. Experimental characterization of de novo proteins and their unevolved random-sequence counterparts. Nat. Ecol. Evol. 7, 570–580 (2023).
- Matsumoto, G., Kim, S. & Morimoto, R. I. Huntingtin and mutant SOD1 form aggregate structures with distinct molecular properties in human cells. J. Biol. Chem. 281, 4477–4485 (2006).
- Tiwari, S., Fauvet, B., Assenza, S., De Los Rios, P. & Goloubinoff, P. A fluorescent multi-domain protein reveals the unfolding mechanism of Hsp70. Nat. Chem. Biol. 19, 198–205 (2023).
- Schultz, T., Martinez, L. & de Marco, A. The evaluation of the factors that cause aggregation during recombinant expression in E. coli is simplified by the employment of an aggregation-sensitive reporter. Microb. Cell Factories 5, 28 (2006).
- Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
- Bushuiev, A. et al. Revealing data leakage in protein interaction benchmarks. Preprint at https://doi.org/10.48550/arXiv.2404.10457 (2024).
- The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2025. Nucleic Acids Res. 53, D609–D617 (2025).
- Nugnes, M. V. et al. DisProt in 2026: enhancing intrinsically disordered proteins accessibility, deposition, and annotation. Nucleic Acids Res. 54, D383–D392 (2026).
- Chan, H. S. & Bornberg-Bauer, E. Perspectives on protein evolution from simple exact models. Appl. Bioinformatics 1, 121–144 (2002).
- Ptitsyn, O. B. & Volkenstein, M. V. Protein Structures and Neutral Theory of Evolution. J. Biomol. Struct. Dyn. 4, 137–156 (1986).
- Dill, K. A., Ozkan, S. B., Shell, M. S. & Weikl, T. R. The Protein Folding Problem. Annu. Rev. Biophys. 37, 289–316 (2008).
- Kocher, C. D. & Dill, K. A. Origins of life: The Protein Folding Problem all over again? Proc. Natl. Acad. Sci. 121, e2315000121 (2024).
- Chiarabelli, C. et al. Investigation of de novo Totally Random Biosequences, Part II. Chem. Biodivers. 3, 840–859 (2006).
- Goldstein, R. A. The evolution and evolutionary consequences of marginal thermostability in proteins. Proteins 79, 1396–1407 (2011).
- Romero-Romero, M. L. & Garcia-Seisdedos, H. Agglomeration: when folded proteins clump together. Biophys. Rev. 15, 1987–2003 (2023).
- Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
- Mahtha, S. K., Venkadesan, S. & Mohanty, D. Comparative evaluation of the prediction accuracy of AlphaFold and ESMFold for monomeric and dimeric proteins. NAR Genomics Bioinforma. 8, lqag002 (2026).
- Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011).
- Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
- Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon: fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods 14, 417–419 (2017).
- Hekkelman, M. L., Salmoral, D. Á., Perrakis, A. & Joosten, R. P. DSSP 4: FAIR annotation of protein secondary structure. Protein Sci. 34, e70208 (2025).