Phosphoproteomics in Daphnia magna as a tool to decipher molecular mechanisms in ecotoxicological studies
Magdalena V. Wilde^a,b^, Jan B. Stöckl^a^, Miwako Kösters^a^, Marco M. Rupprecht^a^, Julian Brehm^c^, Michael Schwarzer^c^, Kathrin A. Otte^d^, Christian Laforsch^c^, Thomas Fröhlich^a,*^
^a^ Gene Center Munich, Laboratory for Functional Genome Analysis (LAFUGA), LMU München, Feodor-Lynen-Straße 25, 81377 Munich, Germany
^b^ Department of Earth- and Environmental Sciences, Paleontology & Geobiology, LMU München, Richard-Wagner-Str. 10, 80333 München, Germany
^c^ University of Bayreuth, Animal Ecology 1 & Bayreuth Center for Ecology and Environmental Research (BayCEER), Universitätsstraße 30, 95447 Bayreuth, Germany
^d^ Institute of Animal Cell and Systems Biology, Universität Hamburg, Martin-Luther-King-Platz 3, 22339 Hamburg, Germany
* Corresponding author
Correspondence should be addressed to:
Dr. Thomas Fröhlich
Email: thomas.froehlich@lmu.de
LMU Munich
Feodor-Lynen-Str. 25
81377 München
Germany
Keywords: Phosphoproteomics, Daphnia , Ecotoxicoproteomics, Environmental pollution
Abstract
Pollution of aquatic environments poses an increasingly severe threat to ecosystems worldwide, and understanding its molecular consequences for aquatic organisms requires extensive research and the development of advanced analytical tools. Phosphoproteomics can be particularly valuable for this purpose, as shifts in phosphorylation states can serve as early molecular indicators of toxic exposure. The cladoceran Daphnia is a keystone species in aquatic ecosystems, linking lower and higher trophic levels, and is therefore widely used as a model organism in ecotoxicology to study biological consequences of pollution. Here, we present a simple and effective strategy to analyse the phosphoproteome of Daphnia magna , a commonly used Daphnia species in ecotoxicology. Following TiO₂-based phosphopeptide enrichment and LC-MS/MS analysis, we identified a comprehensive dataset of 3,532 phosphorylation sites across 1,329 phosphoproteins. These proteins were especially involved in signaling pathways and cellular structure and the vast majority have not yet been demonstrated in other Daphnia species. In conclusion, our results demonstrate that a straightforward phosphoproteomic LC‑MS/MS workflow in D. magna can serve as a powerful tool for investigating adverse molecular effects caused by anthropogenic pollution, such as microplastics or pharmaceuticals.
Statement of significance
The dataset presented here demonstrates the feasibility of a simple yet effective strategy to perform phosphoprotemics in Daphnia magna , and it will be particularly valuable for future ecotoxicoproteomics research using this model organism.
Introduction
Environmental pollution is one of the biggest challenges of our time. Therefore, a comprehensive understanding of the molecular responses underlying various pollutants (e.g. microplastics, pharmaceuticals, pesticides) is of utmost importance. One of the most important biochemical processes for modulation of protein activity in eukaryotic organisms are posttranslational modifications (PTMs) [1][2]. Protein phosphorylations are reversible and very prominent PTMs and play a key role in intracellular signal transduction and affect nearly all basic cellular biochemical events, such as cell cycle, differentiation, and proliferation [3][4][5]. Changes in protein phosphorylation capture early pathway-level stress signaling and can therefore serve as early indicators of environmental change [6][7]. Phosphoproteomics serves to identify and quantify protein phosphorylations throughout the proteome and provides a better understanding of complex phosphorylation-based signaling networks [4][8][9]. Understanding these phosphorylation-coordinated networks necessitates knowledge of specific amino acid modifications with both spatial and temporal resolution, which remains analytically complex and challenging [10]. For instance, phosphopeptides represent only a small fraction of all peptides present in a cell lysate due to the low stoichiometry of site-specific phosphorylations, and therefore need enrichment before LC-MS/MS measurements, requiring more sample material than standard analyses [4][9]. Although several phosphoproteomic studies have been conducted in the field of ecotoxicology, improving the understanding of the effects of pollutants on organisms [11][12][13][14], to our knowledge, there has only been one study addressing the phosphoproteome of the cladoceran Daphnia . In 2014, Kwon et al. [15] performed the first global screening of phosphoproteins and their phosphorylation sites in Daphnia pulex . As Daphnia are key species of lake and pond ecosystems and are often used in ecotoxicological studies [16][17], deeper insights into their molecular response to contaminants can help to understand the consequences of environmental pollution, especially since freshwater ecosystems suffer from various manmade threats (e.g., plastic pollution, agricultural activities, or sewage) [18]. Here, we present a comprehensive dataset of phosphorylated D. magna proteins that can serve as a reference for future proteomics studies on this organism, whether in an ecotoxicological context or to identify fundamental molecular processes.
Implementing an easy-to-perform phosphoproteomic workflow (Figure 1) on pooled D. magna samples, we were able to identify 3532 unique phosphorylation sites in 1329 phosphoproteins.
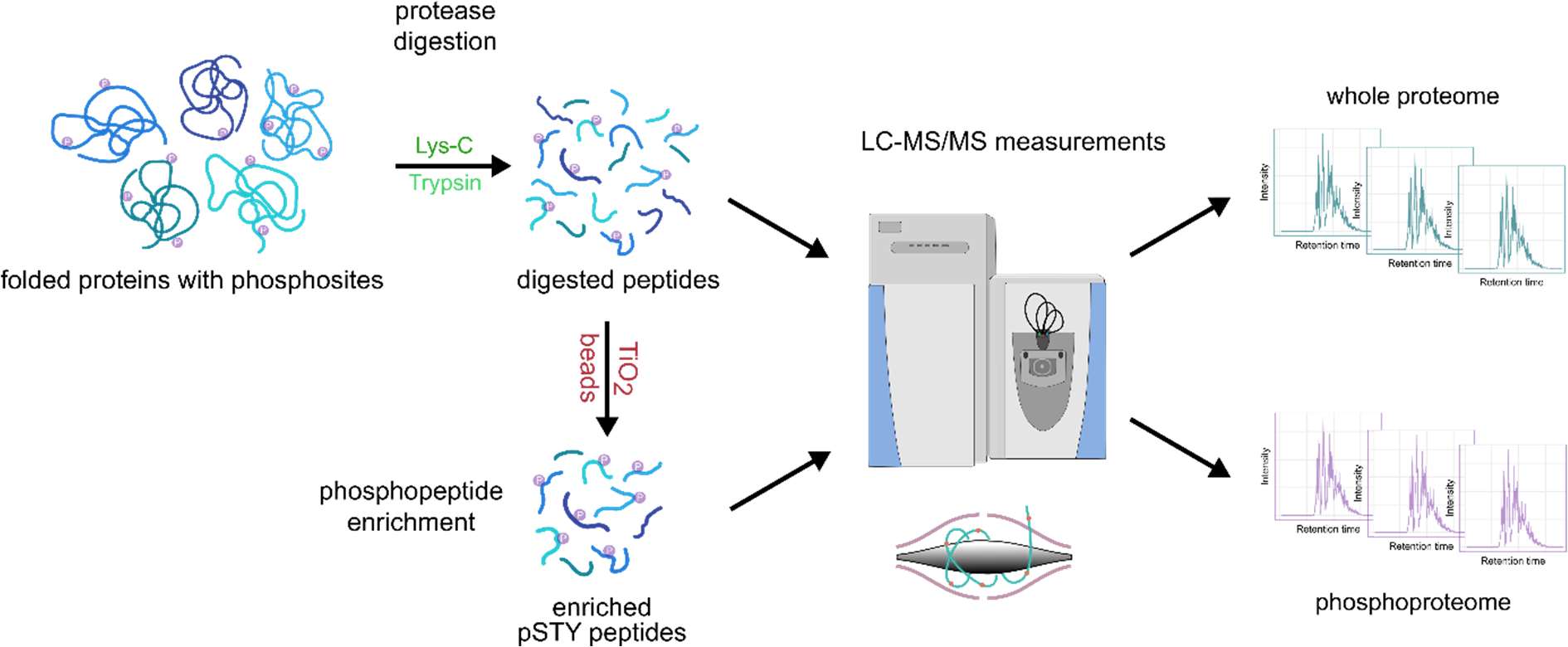
For the first time, we demonstrate a phosphoproteomic LC-MS/MS approach in combination with an in-solution digestion for Daphnia magna , providing an easy-to-perform yet effective method. To obtain sufficient material for phosphoproteomic analysis, 10 adult females of the D. magna clone K34J were pooled in a reaction tube, the remaining water was carefully removed with filter paper and then snap-frozen in liquid nitrogen. The pooled samples (n=5) were homogenized by ultrasonication in 200 µl of lysis buffer (8 M urea, 50 mM ammonium bicarbonate) supplemented with protease inhibitors (one Ultra tablet mini per 10 ml buffer (Roche, Germany)). The protein quantification was done using the Pierce 660 nm Protein Assay (Thermo Fisher Scientific, U.S.A). After reduction and alkylation, sequential digestion, first with Lys-C (4 h, 37 °C) followed by trypsin (overnight, 37 °C), was performed. To obtain optimal conditions for phosphopeptide enrichment, a desalting step was done using the SepPak® Kit (Sep-Pak Vac 1cc (100 mg) tC18 Cartridges, Waters, U.S.A) following the manufacturer’s instructions. For the enrichment of phosphopeptides the High-Select™ TiO₂ Phosphopeptide Enrichment Kit (Thermo Fischer Scientific, U.S.A.) was used. In a final step, enriched phosphopeptides were dried using a vacuum concentrator (Bachofer, Germany).
LC-MS/MS analysis was performed using an Ultimate 3000 RSLC (Thermo Fisher Scientific, U.S.A.) connected to a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, U.S.A.). Phospho-enriched peptides were resuspended in 15 µl 0.1% formic acid (FA), loaded on a trap column (PEP-Map100 C18, 75 µm × 2cm, 3 µm particles (Thermo Fisher Scientific, USA)) and separated on a reversed-phase column (PepMap RSLC C18, 75µm × 50 cm, 2µm particles, Thermo Scientific, U.S.A) at a flow rate of 250 nl/min. A 30-min gradient of 3-25% solvent B followed by 5-min increase to 40% was used. Solvent A consisted of 0.1 % FA, and solvent B of 0.1 % FA in ACN. After separation, the column was washed using 85% solvent B for 10-min. For MS/MS analysis, the data-dependent acquisition method consisted of cycles of one MS scan with a mass range of m/z 300-1600 at a resolution of 60000, followed by a maximum of 15 MS/MS scans at a resolution of 15000. We deposited the mass spectrometry proteomics data to the ProteomeXchange Consortium ( http://proteomecentral.proteomexchange.org ) via the PRIDE partner repository [19] with the project accession: PXD077473.
Raw mass spectrometric data was analyzed using MaxQuant [20] (v. 2.0.3.0) and Perseus [21] (v. 1.6.7.0). In MaxQuant, the Andromeda search engine score filter was left at the default setting of 40, and the enzyme specificity was set to trypsin. Cysteine carbamidomethylation was set as a fixed modification. N-acetylation of protein, methionine oxidation, and the phosphorylation of Serine, Threonine, Tyrosine (STY) were set as variable modifications. For protease digestion, up to two missed cleavages were allowed. The MS/MS spectra were matched against a D. magna UniProt FASTA database, which was run through CD-Hit [22] with a 95% sequence identity cut-off to reduce redundancy. In Perseus, phosphosites were filtered for a localization probability greater than 75%.
Results
In total, we identified 3532 phosphorylation sites (Supplemental Table 1) and 3192 phosphopeptides, which could be assigned to 1329 phosphoproteins, with an FDR of 1%. Besides the study by Kwon et al. [15], in which they reported the first global screening of phosphoproteins in Daphnia pulex , this is, to our knowledge, the second phosphoproteome dataset in Daphnia and one of the most comprehensive phosphoproteomic analyses of aquatic organisms in recent years. Most of the phosphoproteomics studies on higher developed aquatic organisms in the last few years have focused on fish [23][24][25][26] while invertebrates [27][28] have been studied to a lesser extent. Kwon et al. [15] identified 103 phosphorylation sites in 91 proteins in D. pulex . We compared the results of our study with the phosphoproteins identified by Kwon et al. [15] by blasting the identified D. pulex phosphoproteins against our D. magna results. We considered phosphoproteins that had at least 90% query coverage and 90% identity to be shared in both studies. This applied to 39 phosphoproteins, while 52 were not in our dataset, and 1290 are novel identifications (Figure 2A). The intensities of identified phosphoproteins show a dynamic range larger than five orders of magnitude. Gerritsen et al. [29] stated that MS-based phosphoproteomics must be able to handle a large dynamic range of phosphorylations in order to identify and quantify ultra-low level, dynamic phosphorylation events, as well as phosphorylations in high-abundance proteins at high stoichiometry. Furthermore, our data showed that 33.4% of the identified proteins were phosphorylated at a single residue (444 proteins), whereas 66.6% (885 proteins) were phosphorylated at two or more residues (Figure 2B). In more detail, we identified 30.8% of the phosphoproteins having 4 or more phosphorylation sites (410 proteins) and only 2.6% having more than 14 sites (35 proteins). In comparison to a study on mice by Huttlin et al. [30], where 80% of proteins contained multiple phosphorylation sites, with 50% being phosphorylated on four or more residues and 10% on more than 14 sites, our findings indicate fewer phosphorylations per protein. Sebé-Pedrós et al. [31] suggest that the phosphosignaling machinery is more sophisticated in vertebrates than in early diverging organisms or non-animal taxa, based on the greater number of proteins with multiple phosphorylation sites. This suggests that the comparatively lower number observed in our study may reflect an inherently lower phosphorylation level in daphnids. The observed distribution of phosphosites among the S/T/Y residues, with serine being by far the most frequently phosphorylated amino acid (91%), followed by threonine (8%), and tyrosine (1%) being the least phosphorylated, is consistent with several studies [32][33][34][35] (Figure 2C).
![Figure 2: (A) The pie chart depicts phosphoproteins that were previously reported by Kwon et al. 2014 [15] and those that were identified in the present study, as well as those that were identified in both studies. (B) Distribution of the phosphorylation sites per protein over all phosphoproteins. Proteins with 21 or more phosphosites per protein were summarized. © Distribution of phosphosites across serine, threonine, and tyrosine residues.](https://qzydlaimdnpogmrslkea.supabase.co/storage/v1/object/public/manuscripts/figures/upload-1778746911934/fig-02-p6.jpeg)
To gain further insight into the biological functions of the identified phosphoproteins, a functional classification analysis was performed in PANTHER [36][37]. For this, the corresponding D. magna sequences were blasted against D. pulex , using Blast+ [38] from NCBI, and were subsequently filtered for best hits. This output was used in PANTHER and plotted as a treemap using the “treemap” package in R Studio [39] (version 2022.07.2). The three most prominent gene ontology terms in the GO aspect “biological process” are cellular process (45.9%, GO:0009987), metabolic process (29.3%, GO:0008152), and biological regulation (19.9%, GO:0065007) (Figure 3A). For the GO aspect “molecular function”, two GO terms, namely binding (32.0%, GO:0005488) and catalytic activity (23.2%, GO:0003824), are the most represented (Figure 3B). The proteomic landscape is generally highly diverse in different organisms, as stated by Müller et al. [40]. Our findings are consistent with their discovery that proteins involved in metabolic processes constitute one of the most prevalent categories of proteins across all organisms. Furthermore, Humphrey et al. [41] pointed out that phosphorylation plays a crucial role in cellular metabolism, acting as a key switch that links metabolic enzymes into complex signal transduction networks and controls protein function through two main mechanisms: modulation of protein-protein interactions and modification of protein conformation.
We also compared the results of our GO analysis with those of Kwon et al. [15] for D. pulex . Expectably, similar patterns are apparent in several sections. Binding and catalytic activity are also the most prominent GO terms in molecular function. Additionally, we found metabolic process, biological regulation, and response to stimulus to be one of the four main categories in the GO category biological processes. Our most abundant GO term, “cellular process”, is not present in Kwon et al. [15], which could possibly be the result of the higher phosphoproteomic depth of our dataset.
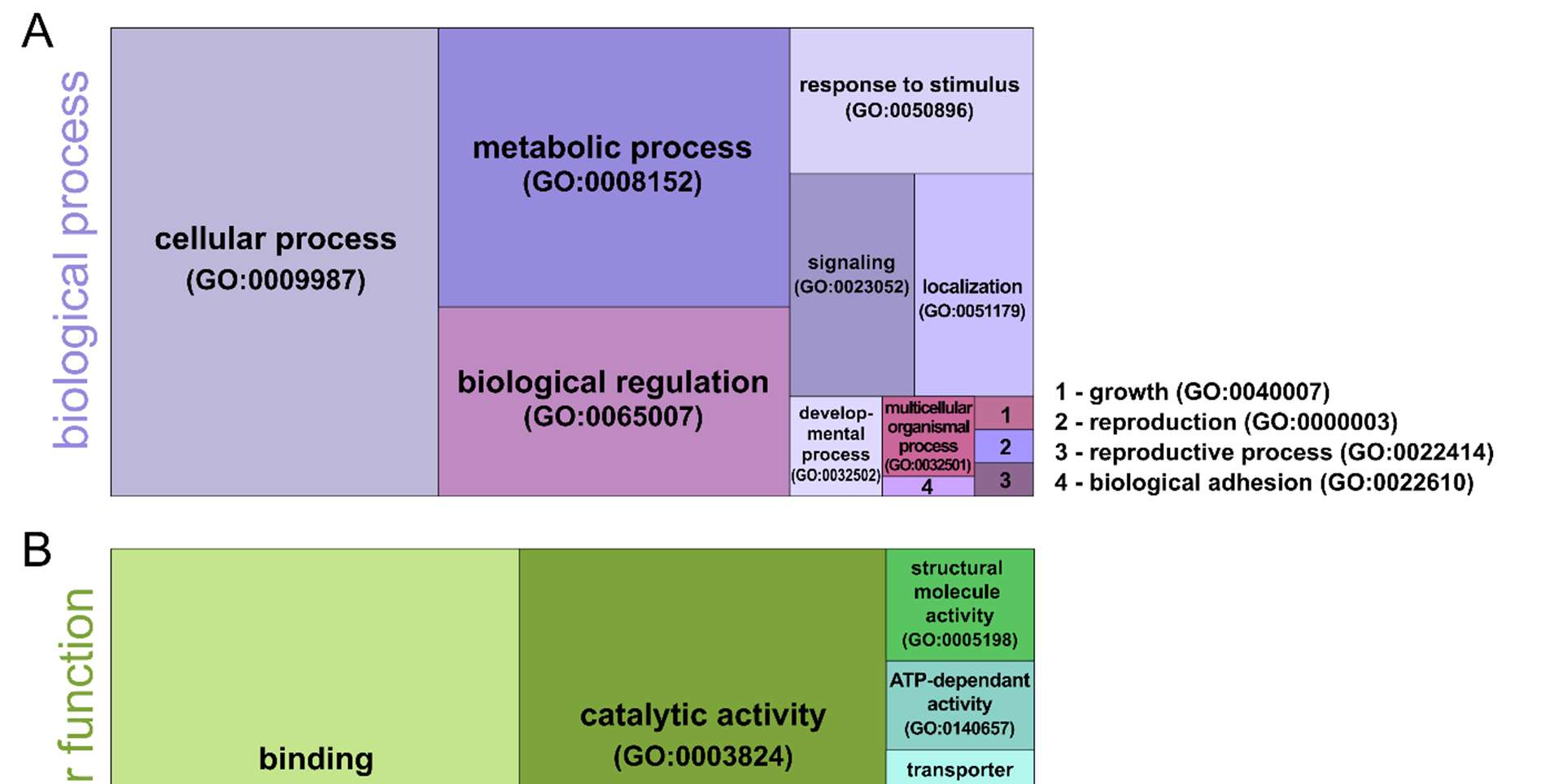
Finally, we used PANTHER to categorize the identified phosphoproteins according to protein classes. Here, we found phosphoproteins assigned to different protein classes, with fundamental RNA metabolism proteins (12.2%, PC00031) and metabolite interconversion enzymes (9.4%, PC00262) being the most abundant ones (Figure 3C). Also, the protein classes cytoskeletal protein (7.2%, PC00085), protein-modifying enzyme (6.6%, PC00260), and translational protein (5.5%, PC00263) were found.
Discussion
In conclusion, our D. magna phosphoproteomic dataset delivers a comprehensive dataset of phosphorylation sites and their corresponding proteins, providing a robust foundation for elucidating regulatory phosphorylation mechanisms in this ecologically important freshwater species. This information is crucial for future studies aimed at deciphering the organism’s response mechanisms to contaminants as well as to environmental changes. The findings from this study are pivotal in advancing our comprehension of ecotoxicological dynamics and in reinforcing the central role of D. magna in ecological research and conservation efforts of freshwater ecosystems.
Acknowledgments
This study was funded by the Deutsche Forschungsgemeinschaft - Project number 391977956 - SFB 1357 to TF and C.L.
Conflict of Interest
The authors declare no conflict of interest.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the PRIDE repository, dataset identifier PXD077473. Project Name: Phosphoproteomics in Daphnia magna as a tool to decipher molecular mechanisms in ecotoxicological studies.
References
- Walsh, C. T., Garneau‑Tsodikova, S., Gatto Jr, G. J., Protein posttranslational modifications: the chemistry of proteome diversifications. Angewandte Chemie International Edition 2005, 44, 7342-7372.
- Mann, M., Jensen, O. N., Proteomic analysis of post-translational modifications. Nature Biotechnology 2003, 21, 255-261.
- Ubersax, J. A., Ferrell Jr, J. E., Mechanisms of specificity in protein phosphorylation. Nature Reviews Molecular Cell Biology 2007, 8, 530-541.
- Macek, B., Mann, M., Olsen, J. V., Global and Site-Specific Quantitative Phosphoproteomics: Principles and Applications. Annual Review of Pharmacology and Toxicology 2009, 49, 199-221.
- Cohen, P., The origins of protein phosphorylation. Nat Cell Biol 2002, 4, E127-130.
- Demertzioglou, M., Antonopoulou, E., Moustaka-Gouni, M., Michaloudi, E., Stress response mechanisms in Daphnia magna : role of HSPs and MAPKs under different environmental conditions. Journal of Plankton Research 2025, 47.
- Silvestre, F., Gillardin, V., Dorts, J., Proteomics to assess the role of phenotypic plasticity in aquatic organisms exposed to pollution and global warming. Integr Comp Biol 2012, 52, 681-694.
- Rigbolt, K. T. G., Blagoev, B., Quantitative phosphoproteomics to characterize signaling networks. Seminars in Cell & Developmental Biology 2012, 23, 863-871.
- Canzler, S., Schor, J., Busch, W., Schubert, K., et al., Prospects and challenges of multi-omics data integration in toxicology. Archives of Toxicology 2020, 94, 371-388.
- Riley, N. M., Coon, J. J., Phosphoproteomics in the Age of Rapid and Deep Proteome Profiling. Analytical Chemistry 2016, 88, 74-94.
- Dash, S., Chandramouli, K. H., Zhang, Y., Qian, P. Y., Effects of poly-ether B on proteome and phosphoproteome expression in biofouling Balanus amphitrite cyprids. Biofouling 2012, 28, 405-415.
- Wang, G., Shen, T., Huang, X., Luo, Z., et al., Autophagy involvement in T lymphocyte signalling induced by nickel with quantitative phosphoproteomic analysis. Ecotoxicology and Environmental Safety 2022, 242, 113878.
- Fang, Y., Deng, X., Lu, X., Zheng, J., et al., Differential phosphoproteome study of the response to cadmium stress in rice. Ecotoxicology and Environmental Safety 2019, 180, 780-788.
- Wang, J., Wang, X., Zhang, C., Zhou, X., Microplastics induce immune suppression via S100A8 downregulation. Ecotoxicol Environ Saf 2022, 242, 113905.
- Kwon, O. K., Sim, J., Yun, K. N., Kim, J. Y., Lee, S., Global Phosphoproteomic Analysis of Daphnia pulex Reveals Evolutionary Conservation of Ser/Thr/Tyr Phosphorylation. Journal of Proteome Research 2014, 13, 1327-1335.
- Altshuler, I., Demiri, B., Xu, S., Constantin, A., et al., An integrated multi-disciplinary approach for studying multiple stressors in freshwater ecosystems: Daphnia as a model organism. Integr Comp Biol 2011, 51, 623-633.
- Miner, B. E., De Meester, L., Pfrender, M. E., Lampert, W., Hairston, N. G., Jr., Linking genes to communities and ecosystems: Daphnia as an ecogenomic model. Proc Biol Sci 2012, 279, 1873-1882.
- Amoatey, P., Baawain, M. S., Effects of pollution on freshwater aquatic organisms. Water Environment Research 2019, 91, 1272-1287.
- Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., et al., The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019, 47, D442-D450.
- Tyanova, S., Temu, T., Cox, J., The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 2016, 11, 2301-2319.
- Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., et al., The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 2016, 13, 731-740.
- Huang, Y., Niu, B., Gao, Y., Fu, L., Li, W., CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680-682.
- Yang, C., Fan, H., Ge, L., Ma, Q., et al., Comparative analysis of quantitative phosphoproteomics between two tilapias ( Oreochromis niloticus and Oreochromis aureus ) under low-temperature stress. PeerJ 2023, 11, e15599.
- Gao, Y., Lee, H., Kwon, O. K., Cheng, Z., et al., Profiling of Histidine Phosphoproteome in Danio rerio by TiO2 Enrichment. PROTEOMICS 2019, 19, 1800471.
- Smith, L. C., Lavelle, C. M., Silva-Sanchez, C., Denslow, N. D., Sabo-Attwood, T., Early phosphoproteomic changes for adverse outcome pathway development in the fathead minnow ( Pimephales promelas ) brain. Scientific Reports 2018, 8.
- Qin, G., Xu, D., Lou, B., Chen, R., et al., iTRAQ-based quantitative phosphoproteomics provides insights into the metabolic and physiological responses of a carnivorous marine fish ( Nibea albiflora ) fed a linseed oil-rich diet. Journal of Proteomics 2020, 228, 103917.
- Shi, H., Wang, J., Liu, F., Hu, X., et al., Proteome and phosphoproteome profiling reveals the regulation mechanism of hibernation in a freshwater leech ( Whitmania pigra ). Journal of Proteomics 2020, 229, 103866.
- Yin, C., Sun, Z., Ji, C., Li, F., Wu, H., Toxicological effects of tris(1,3-dichloro-2-propyl) phosphate in oyster Crassostrea gigas using proteomic and phosphoproteomic analyses. Journal of Hazardous Materials 2022, 434, 128824.
- Gerritsen, J. S., White, F. M., Phosphoproteomics: a valuable tool for uncovering molecular signaling in cancer cells. Expert Rev Proteomics 2021, 18, 661-674.
- Huttlin, E. L., Jedrychowski, M. P., Elias, J. E., Goswami, T., et al., A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell 2010, 143, 1174-1189.
- Sebé-Pedrós, A., Marcia, Capella-Gutiérrez, S., Antó, M., et al., High-Throughput Proteomics Reveals the Unicellular Roots of Animal Phosphosignaling and Cell Differentiation. Developmental Cell 2016, 39, 186-197.
- Sharma, K., D’Souza, Rochelle C. J., Tyanova, S., Schaab, C., et al., Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Reports 2014, 8, 1583-1594.
- Huang, H., Fu, Y., Zhang, Y., Peng, F., et al., Dissection of Anti-tumor Activity of Histone Deacetylase Inhibitor SAHA in Nasopharyngeal Carcinoma Cells via Quantitative Phosphoproteomics. Frontiers in Cell and Developmental Biology 2020, 8.
- Han, R., Wei, Y., Xie, Y., Liu, L., et al., Quantitative phosphoproteomic analysis provides insights into the aluminum-responsiveness of Tamba black soybean. PLOS ONE 2020, 15, e0237845.
- Zhang, Z., Ke, D., Hu, M., Zhang, C., et al., Quantitative phosphoproteomic analyses provide evidence for extensive phosphorylation of regulatory proteins in the rhizobia–legume symbiosis. Plant Molecular Biology 2019, 100, 265-283.
- Thomas, P. D., Ebert, D., Muruganujan, A., Mushayahama, T., et al., PANTHER: Making genome‑scale phylogenetics accessible to all. Protein Science 2022, 31, 8-22.
- Mi, H., Muruganujan, A., Huang, X., Ebert, D., et al., Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 2019, 14, 703-721.
- Camacho, C., Coulouris, G., Avagyan, V., Ma, N., et al., BLAST+: architecture and applications. BMC Bioinformatics 2009, 10, 421.
- R-Core-Team, R: A language and environment for statistical computing. R Foundation for Statistical Computing,Vienna, Austria: 2014. R A Lang. Environ. Stat. Comput. 2018.
- Müller, J. B., Geyer, P. E., Colaço, A. R., Treit, P. V., et al., The proteome landscape of the kingdoms of life. Nature 2020, 582, 592-596.
- Humphrey, S. J., James, D. E., Mann, M., Protein phosphorylation: a major switch mechanism for metabolic regulation. Trends in Endocrinology & Metabolism 2015, 26, 676-687.